
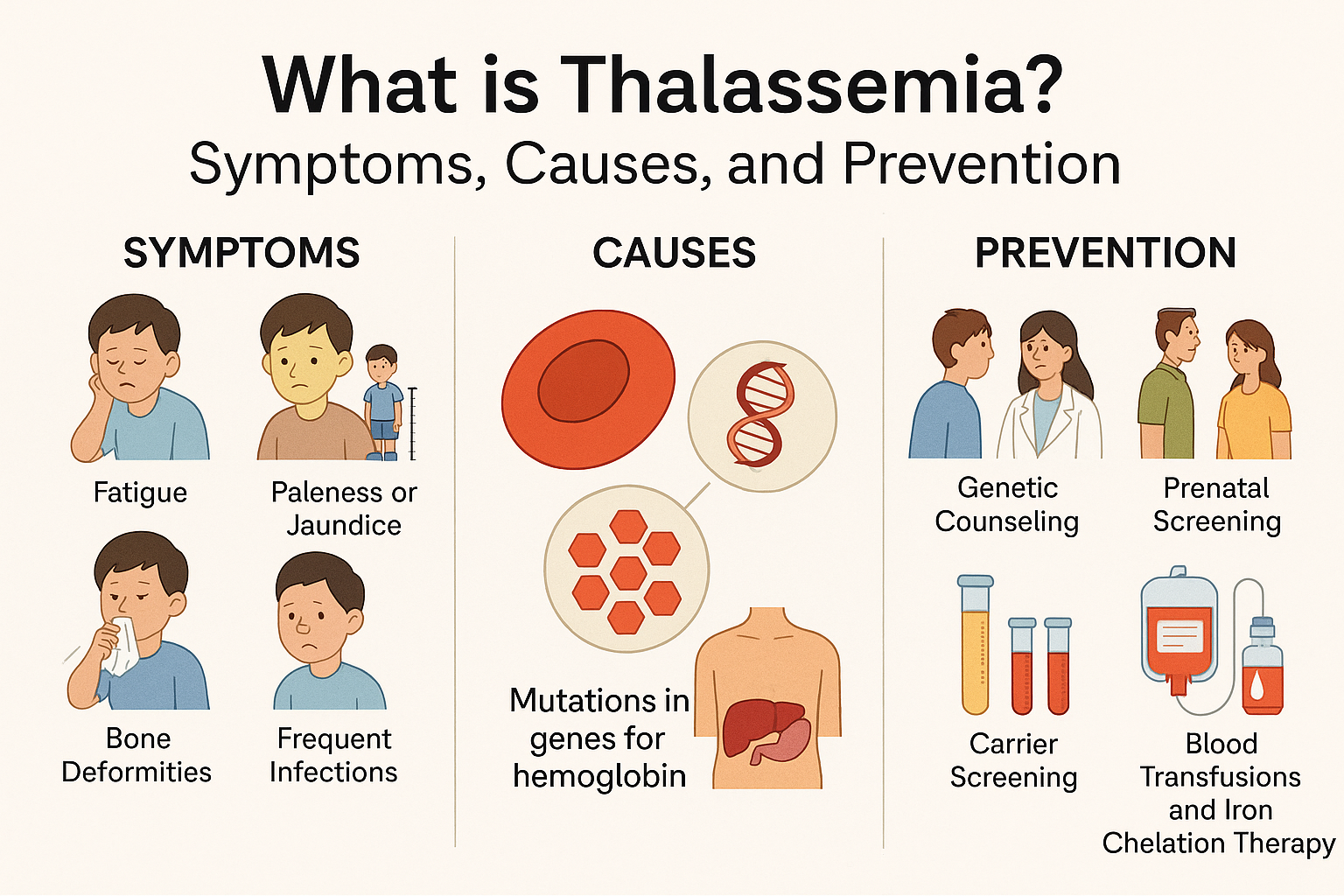
 Thalassemia is a group of inherited blood disorders that affect the body’s ability to produce hemoglobin, a vital protein found in red blood cells. Hemoglobin is responsible for carrying oxygen from the lungs to all parts of the body. When hemoglobin production is reduced or abnormal, the body does not get enough oxygen, leading to a condition called anemia.
Thalassemia is a group of inherited blood disorders that affect the body’s ability to produce hemoglobin, a vital protein found in red blood cells. Hemoglobin is responsible for carrying oxygen from the lungs to all parts of the body. When hemoglobin production is reduced or abnormal, the body does not get enough oxygen, leading to a condition called anemia.
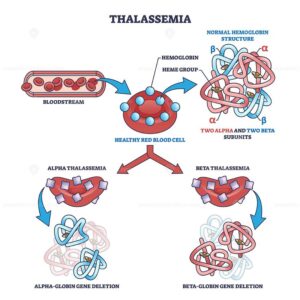
Thalassemia is caused by genetic mutations that are passed from parents to their children. These mutations affect the genes responsible for producing hemoglobin chains. Normally, hemoglobin is made up of two types of protein chains: alpha and beta. Depending on which chain is affected, thalassemia is classified into two main types: alpha thalassemia and beta thalassemia.
Alpha thalassemia occurs when there is a problem with the genes that produce alpha globin chains. A person inherits four genes related to alpha globin, two from each parent. The severity of the condition depends on how many of these genes are affected. If only one gene is affected, the person may not show any symptoms and is called a silent carrier. If two genes are affected, mild anemia may occur. If three genes are affected, the condition becomes more serious and is known as hemoglobin H disease. If all four genes are affected, it can result in a severe condition called hydrops fetalis, which is often fatal before or shortly after birth.
Beta thalassemia occurs when there is a defect in the genes that produce beta globin chains. A person inherits two beta globin genes, one from each parent. If one gene is affected, the person has beta thalassemia minor (also called trait), which usually causes mild anemia or no symptoms at all. If both genes are affected, it results in beta thalassemia major, a severe form of the disease that requires regular medical treatment. There is also an intermediate form called beta thalassemia intermedia, which falls between minor and major in severity.
The symptoms of thalassemia vary depending on the type and severity. People with mild thalassemia may not notice any symptoms. However, those with moderate to severe forms often experience fatigue, weakness, pale or yellowish skin, shortness of breath, and dizziness. Children with severe thalassemia may have delayed growth and development. In some cases, bones may become weak or deformed, especially in the face and skull, due to increased bone marrow activity as the body tries to produce more red blood cells.
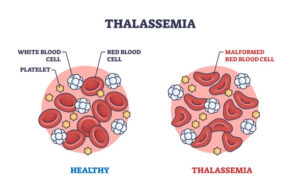
Another common problem in thalassemia is an enlarged spleen. The spleen is an organ that helps filter blood and remove damaged red blood cells. In thalassemia, because many red blood cells are abnormal, the spleen works harder and becomes enlarged. This can further worsen anemia and may require surgical removal of the spleen in some cases.
Diagnosis of thalassemia is usually done through blood tests. A complete blood count (CBC) can show low hemoglobin levels and smaller-than-normal red blood cells. Additional tests such as hemoglobin electrophoresis help identify the type of hemoglobin present in the blood and confirm the diagnosis. Genetic testing may also be used to detect specific mutations. In many countries, including India, screening programs are encouraged, especially for couples planning to have children.
Treatment for thalassemia depends on the severity of the condition. Mild cases may not require any treatment. For moderate to severe cases, regular blood transfusions are often needed to maintain healthy hemoglobin levels. However, frequent transfusions can lead to a buildup of iron in the body, known as iron overload. This can damage organs such as the heart, liver, and endocrine glands. To prevent this, patients are given iron chelation therapy, which helps remove excess iron from the body.
In some cases, bone marrow or stem cell transplantation may be considered as a potential cure. This procedure involves replacing the patient’s defective bone marrow with healthy marrow from a compatible donor. While it can be effective, it also carries risks and is not suitable for everyone.
Living with thalassemia requires ongoing medical care and monitoring. Patients need regular check-ups, blood tests, and sometimes imaging studies to assess organ function. A healthy lifestyle, including a balanced diet and proper medical care, can help manage the condition. It is also important to avoid unnecessary iron supplements unless prescribed by a doctor, as excess iron can worsen complications.
Prevention plays a crucial role in reducing the impact of thalassemia. Since it is a genetic disorder, genetic counseling is recommended for individuals who are carriers or have a family history of the condition. If both parents are carriers, there is a 25 percent chance with each pregnancy that the child will have severe thalassemia. Prenatal testing can help detect the condition early in pregnancy and allow families to make informed decisions.
Thalassemia is more common in certain parts of the world, including South Asia, the Mediterranean region, the Middle East, and Southeast Asia. In India, it is considered a significant public health concern, with thousands of children born each year with severe forms of the disease. Awareness programs, early screening, and proper medical care can greatly improve outcomes and quality of life for affected individuals.
In conclusion, thalassemia is a lifelong genetic blood disorder that affects hemoglobin production and leads to anemia. While mild forms may have little impact, severe forms require regular treatment and careful management. Advances in medical science, including improved transfusion techniques and stem cell transplantation, have significantly increased life expectancy for patients. With proper awareness, early diagnosis, and preventive measures, the burden of thalassemia can be reduced, and those affected can lead healthier lives.
Types of Thalassemia
1. Alpha Thalassemia
Caused by defects in alpha globin genes
Severity depends on how many genes are affected (1 to 4)
1 gene: Silent carrier (no symptoms)
2 genes: Mild anemia
3 genes: Moderate to severe (Hemoglobin H disease)
4 genes: Very severe (usually fatal before birth)
2. Beta Thalassemia
Caused by defects in beta globin genes
Types:
Minor (trait): Mild or no symptoms
Intermedia: Moderate symptoms
Major: Severe, needs lifelong treatment
 Causes
Causes
Inherited from both parents (major) or one parent (minor)
If both parents are carriers:
25% chance: Normal child
50% chance: Carrier
25% chance: Severe thalassemia
 Symptoms
Symptoms
Mild cases:
Slight weakness
Mild anemia
Often no symptoms
Moderate to severe cases:
Fatigue and weakness
Pale or yellowish skin
Shortness of breath
Dizziness (giddiness)
Slow growth in children
Bone deformities (especially face)
Enlarged spleen
Dark urine